Single Cell Genomics Facility
The Single Cell Genomics Facility is funded by the CRUK City of London Centre based at the UCL Cancer Institute. The facility provides a streamlined service from library prep to bioinformatic analysis. The facility currently has the necessary equipment to run samples on the 10X Genomics Chromium Controller and Fluidigm C1 platforms. We are able to run single-cell RNA-seq, ATAC-seq, CITE-seq experiments and are open to discussions about adding other methods you may be interested in. Users will provide the cell suspension to the facility which is immediately run on the desired platform followed by library preparation and sequencing on an Illumina platform. Quality control, visualisation of cell populations and differential gene expression analysis can then be carried out by the in-house single cell bioinformatician. Bespoke analyses to fit the unique nature of the data is also part of the service.

Contact


Members of the UCL Cancer Institute and CRUK City of London Major Centre are allowed to use the facility. City of London Centre members outside of UCL (Kings, Barts and Crick) won’t have direct access to use the 10X Chromium but can submit samples to the facility to be run by the technician.
Yes. UCL staff and students who are not based in the Cancer Institute can have access to use the 10X Genomics Chromium Controller (if booked in advance on Clustermarket) but only after receiving relevant training on using the instrument. You are then able run the sample on the instrument and can perform the library prep back in your lab.
Users must provide the facility with the cell suspension, relevant 10X Genomics kits and Illumina sequencing kits. All samples are currently being processed on the NextSeq. A small fee will be charged to users for additional reagents and consumables required to process the samples which are not provided with the 10X Genomics and Illumina kits. There is no charge for the bioinformatic analysis.
Turnaround times will be decided on an experiment by experiment basis. This is dependent on workload, annual leave, other commitments and bookings of sequencers. Please get in touch as soon as possible if you want to use the facility and get an estimated turnaround time.
This is dependent on how many cells you have and are interested in recovering. For a typical recovery of 10,000 cells, we aim to load 20,000 cells into the 10X Chromium Controller. If these cells are being sorted beforehand, we recommend recovering around 60,000 cells on the sorter before submitting to the facility.
- Single Cell Gene Expression
- Single Cell Immune Profiling
- Single Cell ATAC
- CITE-Seq (quantification of proteins with RNA)
This method measures gene expression (mRNA abundance) in single cells. Because of this, the technique can reveal heterogeneity within a cell population, identifying and characterising rare cell types without prior knowledge of cell subtypes or cell markers. You can define novel cell types and cell states and discover new biomarkers for specific cell subpopulations. You can analyse and understand cellular heterogeneity and see how this contributes to the biological system you are interested in.
An example of a clustering and gene expression analysis performed by the single cell genomics facility bioinformatician is shown in Figure 1.
Publications using Single Cell Gene Expression techniques can be found here: https://www.10xgenomics.com/resources/publications/?solution=single-cell
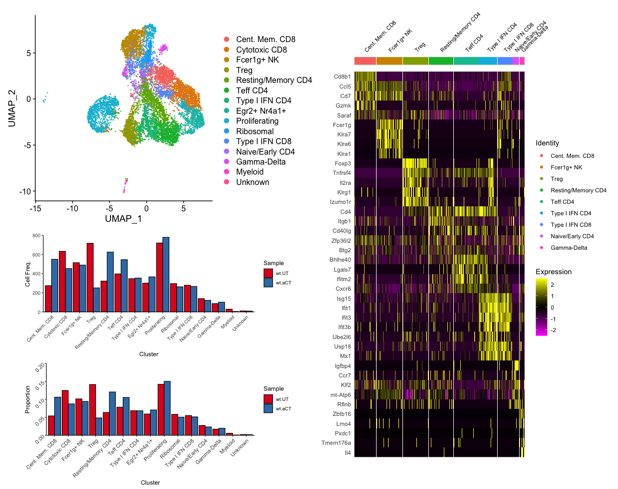
Figure 1. Determining T cell subtypes present in a mouse tumor model, and comparative analysis of T cell population changes between untreated (wt.UT) and ⍺-Ctla4 treated (wt.aCT) tumor microenvironments.
This approach enables the user to assay the TCR or immunoglobulin chain and expression profile of each immune cell in their sample simultaneously. Clonality and repertoire diversity can then be assessed in the context of immune subtypes for tens of thousands of cells.
Feature barcoding technology uses barcode conjugated antibodies which can bind specific surface markers, and can be applied in parallel to the immune profiling approach. This enables additional profiling of antigen specificity or key cell surface molecules.
An example of a TCR clonality analysis performed by the single cell genomics facility bioinformatician is shown in figure 2.
Publications using Single Cell Immune Profiling techniques can be found here: https://www.10xgenomics.com/resources/publications/?solution=vdj
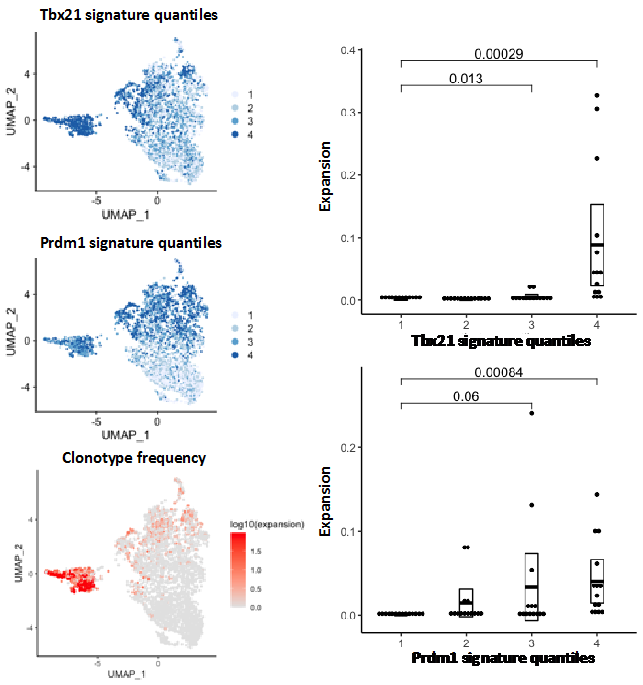
Figure 2. TCR clonality analysis of non-Treg CD4 cells from non-small lung cancer data (Guo et al 2018) showing correlation between TCR expansion and Tbx21 and Prdm1 gene expression signatures
The Single Cell Assay for Transposase Accessible Chromatin (ATAC) solution allows you to map the epigenetic landscape at a single cell resolution. Open chromatin regions can be identified, as the technique uses a transposase enzyme to preferentially tag accessible DNA regions with sequencing adaptors, which enable you to create a sequencing ready library. Tens of thousands of cells can be analysed to understand the epigenetic and regulatory variations.
The technique allows you to investigate and compare open chromatin in regions of interest and cluster and identify cells based on open chromatin regions and enriched transcription factor (TF) motifs.
Further publications using Single Cell ATAC Seq techniques can be found here: https://www.10xgenomics.com/resources/publications?page=1&sortBy=publications-relevance&query=&refinementList%5BproductGroups%5D%5B0%5D=Single%20Cell%20ATAC
Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-Seq) uses DNA-barcoded antibodies to convert detection of proteins into a quantitative sequence-able readout. Antibody-bound oligos act as synthetic transcripts that are captured during the 10X Genomics Gene Expression protocol. This allows for immune-phenotyping of cells with a potentially limitless number of markers and unbiased transcriptome analysis using the 10X Genomics approach.
Further publications using Single Cell CITE-Seq techniques can be found here: https://cite-seq.com/publications/
Sequencing Depth per Experiment
- 3ʹ Gene Expression Library – Minimum 20,000 read pairs per cell
- 5′ Gene Expression library – Minimum 20,000 read pairs per cell
- 3’ Cell Surface Protein Library – Minimum 5,000 read pairs per cell
- 5’ V(D)J Enriched library – Minimum 5,000 read pairs per cell
- 5’ Cell Surface Protein library – Minimum 5,000 read pairs per cell
- Single Cell ATAC – Minimum 25,000 read pairs per nucleus
- CITE-Seq – Minimum 20,000 read pairs per cell
Everyone with access to the Single Cell Genomics Facility (as outlined above) also has access to the bioinformatics facility. Advice on platform, cell numbers, cell health and other factors which can affect downstream analysis can be provided prior to running your experiment. Once the sequenced data are retrieved, an entire bioinformatics pipeline can be provided by the facility, complete with bespoke analysis based on feedback and requirements from the user.
Users can also request analysis on data that has not been generated by the facility. A large amount of single-cell data is available from databases such as NCBI GEO and SRA. Novel insights from these datasets could be the basis for preliminary data for a grant proposal or form a complete meta-analysis study. Table 1 lists several scRNA-seq datasets that are available as user-friendly interactive webpages.

Table 1. Publicly available interactive T cell scRNA datasets
We are always looking to add new techniques and instruments into the facility. Please get in touch to find out about the latest services we provide and if you have any suggestions on services/ instruments you would like us to provide so we can explore the options.